

第一作者:Jianchao Dong
通訊作者:Xuyong Yang & Ning Wang
通訊單位:上海大學,吉林大學
DOI:https://doi.org/10.1038/s41565-024-01852-6
研究背景
盡管在綠色和紅色金屬鹵化物鈣鈦礦發光二極管(PeLEDs)方面取得了重大進展,但藍色PeLEDs,尤其是深藍色,由于嚴重的相分離導致的電致發光光譜偏移以及在寬帶隙鈣鈦礦發射極中低激子利用率,仍然大大落后。
本文亮點
本文提出了一種多價固定策略,通過引入一種含氟氧多原子分子來實現高效且光譜穩定的深藍色PeLEDs。系統的實驗和廣泛的5000fs從頭分子動力學模擬揭示,多價效應的關鍵在于氫鍵(F···H–N)、離子鍵(F–Pb)和配位鍵(C=O:Pb)與鈣鈦礦的三種相互作用,它們協同穩定鈣鈦礦相并增強激子的輻射復合。深藍色鈣鈦礦發射器的激子濃度和激子復合率分別增加了1.66倍和1.64倍。本文的目標PeLEDs在459nm的深藍色發射波長下顯示出高達15.36%的峰值外量子效率,并且在0.45 mA cm−2的恒定電流密度下壽命達144分鐘。此外,深藍色PeLEDs在穩定的驅動電流下保持了恒定的光譜峰值,其CIE色品坐標為(0.136, 0.051),持續時間為60分鐘。
圖文解析
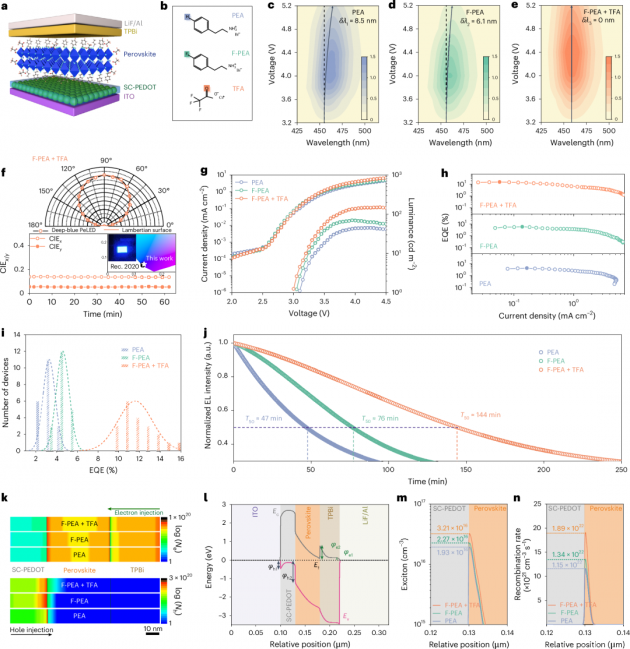
圖1| 深藍色PeLEDs的器件性能和自洽光電模擬
要點:
1.本文制造了深藍色PeLEDs,其器件結構如下:涂有氧化銦錫(ITO)的玻璃基底/摻雜檸檬酸鈉的聚(3,4-乙撐二氧噻吩)聚苯乙烯磺酸鹽(SC-PEDOT)作為空穴傳輸層,該層可以同時提高PEDOT:PSS層的空穴注入能力和形貌,如本研究團隊之前的工作所報道的那樣(圖1a)。RDP發光層通過旋涂前驅體溶液并使用抗溶劑(間二甲苯)輔助的方法沉積。圖1b展示了三種有機分子的結構,分別基于(苯乙基溴化銨)PEA、(4-氟苯乙基溴化銨)F-PEA和(銫三氟乙酸鹽)TFA。所有器件均展現出深藍色電致發光(EL)。然而,在電壓從3.2V增加到5.2V時,PEA處理的器件中觀察到EL紅移8.5納米(圖1c),而F-PEA處理的器件也顯示出輕微的峰值偏移(6.1納米)(圖1d)。相比之下,F-PEA+TFA處理的器件完全沒有EL峰值偏移(圖1e),且峰值EL亮度高于PEA處理和F-PEA處理的器件。
2.F-PEA+TFA處理的PeLED的角發射強度遵循朗伯分布(圖1f),并且CIE值可以在連續工作條件下保持不變。圖1f的插圖展示了一個在3.7V電壓下工作的PeLED的生動照片,它發出明亮的深藍色光,CIE色坐標為(0.136, 0.051),與Rec. 2020規定的深藍色標準(0.131, 0.046)非常匹配。目標最佳性能的PeLED在459納米處顯示出高達15.36%的峰值外量子效率(EQE)(圖1g,h),這在深藍色PeLED領域大大超過了以往的工作。EQE再現性的PeLEDs與TFA也呈現在直方圖中(圖1i),平均EQE為11.56±1.7%,是PEA處理器件的三倍。
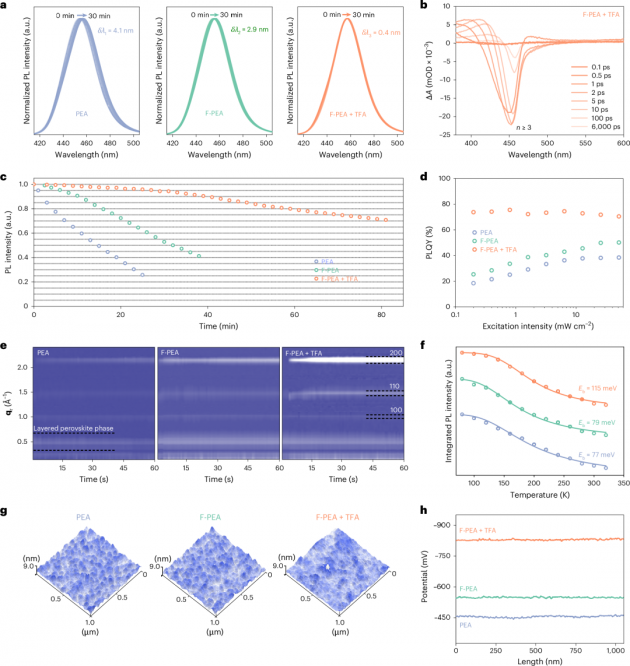
圖2| RDP膜的光學和電學特性
要點:
1.本文評估了RDP薄膜的發射穩定性,以探索其在電致發光(EL)光譜特性上的改進。在80℃的加熱條件下,本文記錄了RDP薄膜的光致發光(PL)峰位(圖2a)和PL強度(圖2c)。觀察到用PEA處理的RDP薄膜遭受嚴重的熱猝滅和光學遷移。然而,用F-PEA處理的RDP薄膜顯示出改善的穩定性,而F-PEA+TFA處理的RDP即使連續加熱1小時后,仍保持其初始發射強度的80%以上。這是PEA處理的RDP保持其初始發射強度時間的十倍。為了深入了解RDP薄膜的量子阱寬度分布和光學性質,進行了瞬態吸收光譜分析。PEA和F-PEA樣品表現出多n相分布,包括n=2的鈣鈦礦相。相比之下,在引入TFA后,F-PEA+TFA樣品中的n=2區域明顯受到抑制(圖2b)。F-PEA+TFA樣品的長組分1,792 ps表明非輻射復合路徑減少,激發載流子被有效利用。
2.本文進一步通過穩態PL和紫外-可見吸收光譜分析了發光特性。發光強度的增加表明F-PEA和TFA分子對RDP薄膜具有優異的鈍化效果。更高的PLQYs和更長的PL壽命也證實了缺陷密度的降低。此外,F-PEA+TFA處理薄膜的PLQYs顯示出較弱的激發強度依賴性(圖2d),表明其光學特性優越。而且,從PEA處理到F-PEA處理再到F-PEA+TFA處理的RDP樣品,激子結合能(Eb)從77 meV增加到115 meV(圖2f),光學聲子能量(?ω)從47.52 meV增強到79.35 meV。這些結果表明,F-PEA+TFA處理的RDP經歷了強烈的限制效應和熱抗猝滅效應,這與PLQYs和熱穩定性測量結果一致。有趣的是,鈣鈦礦的晶粒尺寸沒有明顯變化,而RDP薄膜的形態在加熱前后有所不同。晶體在PEA衍生的RDP薄膜表面沉淀,而在F-PEA衍生的RDP薄膜上形成孔洞,表明加熱時有機胺的損失。相比之下,在F-PEA+TFA衍生的RDP薄膜中可以清楚地看到晶粒的顯著融合,同時基本上保持了薄膜的完整性。本文推測TFA與RDPs之間存在多價效應(多個化學鍵),導致有機陽離子緊密結合在鈣鈦礦表面上,增加了激子結合以及提高了鈣鈦礦結構的穩定性。
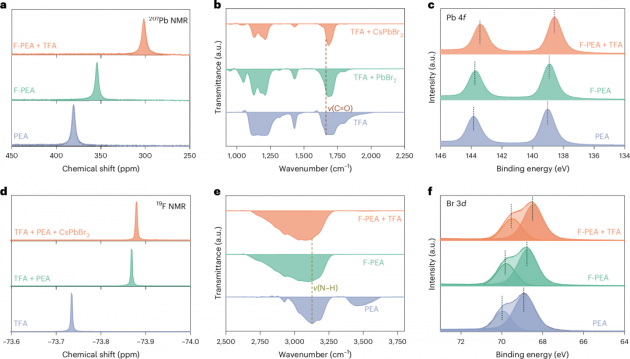
圖3| 有機物與RDPs之間的多價效應
要點:
1.為了研究RDPs中的多價效應,本文進行了核磁共振(NMR)實驗,以檢測有機物與RDPs之間的強相互作用。對于F-PEA+TFA處理的RDP樣品,207Pb的化學位移位于301.7 ppm,明顯偏離了F-PEA處理(354.4 ppm)和PEA處理(380.7 ppm)的樣品(圖3a)。本文將這種偏移歸因于F和Pb之間形成的離子鍵(F–Pb),或者羰基(C=O)中氧原子上的孤電子對向Pb2+離子的空6p軌道捐贈。進行了反射微傅里葉變換紅外(FTIR)光譜和X射線光電子能譜(XPS)實驗,以觀察C=O伸縮振動(圖3b)和Pb 4f的信號(圖3c)。在TFA粉末的FTIR光譜中,來自C=O伸縮振動(ν(C=O))的峰位于1665 cm−1,在與CsPbBr3粉末混合后,波數移至更高的1682 cm−1,表明TFA與Pb2+陽離子之間的配位。
2.與PEA處理的RDP相比,F-PEA處理和F-PEA+TFA處理的RDP顯示出Pb 4f核心能級的峰向較低的結合能偏移,進一步驗證了配位鍵(C=O:Pb)的形成。從PEA處理的RDP到F-PEA+TFA處理的RDP,Pb 4f7/2的光電子發射能移(0.4 eV)從139.0 eV移至138.6 eV,與F-PEA處理的RDP相比,表明C=O:Pb的相互作用占主導。此外,由于C=O:Pb的配位鍵,Br 3d和Cl 2p(圖3f)也顯示出向較低結合能的偏移,導致Pb2+與鹵素陰離子之間的靜電相互作用發生變化。這些結果提供了證據,表明TFA鈍化有效減少了由離子空位引起的非輻射復合。
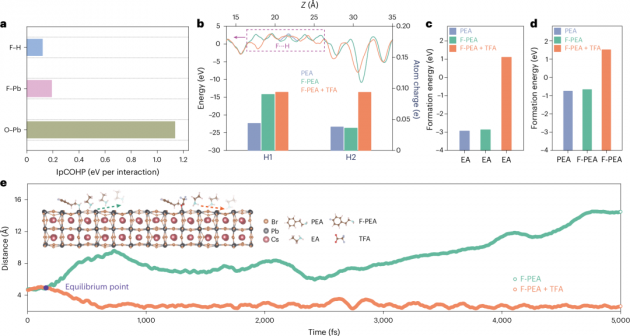
圖4| 通過多價效應固定深藍色鈣鈦礦發射體的機制
要點:
1.本文使用密度泛函理論(DFT)計算進一步闡明了多價效應導致的固定機制。計算結果證實,Pb離子與周圍的F和O離子有明顯的電荷轉移。通過DFT模擬引入晶體軌道哈密頓布居數(COHP)來量化和比較TFA和RDP之間的不同作用力(圖4a)。觀察到的O和Pb之間的最大作用力表明其主導相互作用,其次是F和Pb,以及F和H。這些結果與本文實驗發現一致。有無有機組分的差異電荷密度分布圖和電子定域函數清楚地展示了TFA分子與RDPs之間的相互作用。
2.本文對三種結構進行靜電勢表征,以揭示電荷分布并確定可能的相互作用。計算顯示,氟與其周圍的氫原子之間有明顯的電荷轉移(圖4b),這是由于氫鍵的形成。因此,有機組分的空位形成能增加(圖4c,d),說明了氟元素對胺的固定作用。此外,鹵素離子的空位形成能也增加了,這與飛行時間二次離子質譜(TOF-SIMS)的結果一致。通過從頭算分子動力學模擬對RDP樣品的動態特性進行了表征。如圖4e所示,在沒有TFA的情況下,有機組分(乙胺(EA)分子)隨時間逐漸從RDP的無機本體中脫離。然而,在TFA處理的RDP情況下,有機組成在模擬過程中直到5000 fs都保持不變。這一重要發現與RDP薄膜的穩定性測試結果一致(圖2a)。
總結與展望
本文證明了使用多價效應策略可以實現來自RDPs的高性能深藍色電致發光(EL),其峰值EQE在459 nm處達到15.36%,并且在穩態電場下具有顯著的光譜穩定性。EL性能的提升歸功于三種化學鍵提供的多重保護,這些化學鍵同時防止了相分離和離子遷移,并增加了激子的輻射復合。本文的實驗結果、理論計算和半導體自洽光電模擬闡明了通過多價效應固定可以全面控制深藍色RDPs的結構、晶體學和光電子性能。這一方法展示了RDPs實現高效且穩定的深藍色PeLEDs的潛力。除了深藍色發射外,所提出的策略很可能廣泛適用于穩定其他顏色鈣鈦礦的晶體結構,為未來全彩顯示技術的發展提供支持。
原文詳情:Dong, J., Zhao, B., Ji, H. et al. Multivalent-effect immobilization of reduced-dimensional perovskites for efficient and spectrally stable deep-blue light-emitting diodes. Nat. Nanotechnol. (2025).
https://doi.org/10.1038/s41565-024-01852-6